In-Silico Charakterisierung von krankheitsassoziierten Bereichen in Ionenkanal- und Transporterproteinen
Ziel von Teilprojekt 3 ist es ein System zu entwickeln, um in-silico spezifische genetische Varianten von Ionenkanälen bewerten und klassifizieren zu können. Mithilfe dieses Klassifizierungssystems werden wir den Effekt genetischer Varianten vorhersagen und Regionen innerhalb der Ionenkanäle identifizieren, welche sich für die Entwicklung therapeutischer Ziele eignen. Das Projekt gliedert sich in folgende Teilprojekte:
1) Entwicklung einer Datenbank für Ionenkanal-Varianten. Hierzu werden wir alle öffentlich und kommerziell verfügbaren sowie anderweitig durch Konsortien zur Verfügung stehenden genetischen Varianten in Ionenkanälen in einer Datenbank zusammenfassen und mit RNA-Expressionsdaten sowie vorhandenen experimentellen Proteinstrukturen sowie Homologiemodellen für die jeweiligen Ionenkanalproteine ergänzen. Zur Validierung unserer Vorhersagen anhand von experimentellen Daten werden wir den Fokus auf die Genfamilien legen, die in weiteren Treat-ION-Projekten untersucht werden, u.a Calciumkanäle (CACNA1x Genfamilie), sowie eine Gruppe der Kaliumkanäle (KCN Genfamilie).
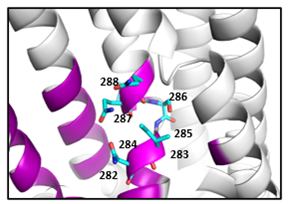
2) Mithilfe von Genfamilien werden wir eine neue Methode für die Interpretation genetischer Varianten entwickeln. Basierend auf einer verbesserten Version einer kürzlich von uns publizierten Methode (Lal et al., 2020 Genome Medicine), werden wir die Konservierung innerhalb von Genfamilien quantifizieren, wodurch evolutionär konservierte Aminosäuren identifiziert werden. Zudem werden wir weitere Informationen, wie beispielsweise die Eigenschaften der Aminosäuren und Informationen aus den Proteinstrukturen, in die neue Methode integrieren, um diese zu optimieren.
3) Korrelationsstudie zwischen den genetischen Varianten, ihrer Position innerhalb von Proteindomänen und den klinischen Phänotypen zur Aufschlüsselung der phänotypischen Heterogenität der Patienten. Hierzu werden wir alle genetischen Varianten zusammen mit den dazugehörigen Patientenphänotypen aus der zentralen Phänotypdatenbank des Projektes (P1) sowie neue Varianten aus den Projekten P4-P8 sammeln. Die komplexen Phänotypen werden anschließend durch Clusteranalysen in symptomatische Untergruppen unterteilt, um dann Korrelationsanalysen zwischen den Untergruppen und der Art, der Position innerhalb des Proteins und den Eigenschaften der genetischen Variante durchzuführen.